Studies have found that when there is a lack of mitochondria in neuron axons, it causes a build-up of proteins, which is a key factor in neurodegenerative diseases. This offers a new target for treatment. Credit: SciTechDaily.com
A significant pathway has been identified for how the reduction of axonal mitochondria disrupts autophagy.
Scientists from Tokyo Metropolitan University have figured out how proteins abnormally accumulate in neurons, which is a feature of neurodegenerative diseases like Alzheimer’s. They used fruit flies to demonstrate that the reduction of mitochondria in axons can directly lead to protein accumulation. At the same time, they found high levels of a protein called eIF2β. Bringing the levels back to normal resulted in a recovery in protein recycling. These findings offer new hope for treatments for neurodegenerative diseases.
The Cellular Factory: Protein Production and Disassembly
Every cell in our bodies is like a busy factory, where proteins are constantly being produced and broken down. Any changes or mistakes in either the production or recycling stages can lead to serious illnesses. Neurodegenerative diseases such as Alzheimer’s and Amyotrophic Lateral Sclerosis (ALS) are known to be accompanied by an abnormal build-up of proteins in neurons. However, the cause of this accumulation remains unknown.
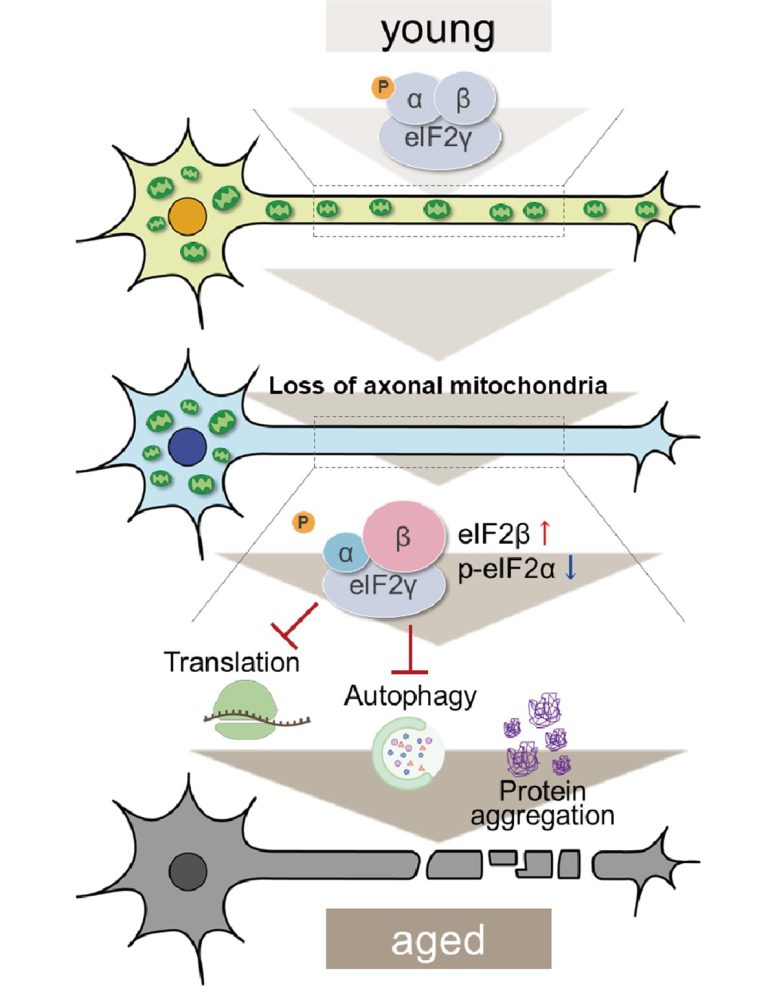
As axonal mitochondria are lost, changes in the parts of eIF2 are observed which hinder translation and autophagy and cause the accumulation of proteins in cells. Credit: Tokyo Metropolitan University
Research Using Drosophila Fruit Flies
A team led by Associate Professor Kanae Ando of Tokyo Metropolitan University has been investigating the reasons for abnormal protein build-up by studying Drosophila fruit flies, a frequently studied model organism that shares many important similarities with human physiology.
They focused on the presence of mitochondria in axons, the long tendril-like extensions that stretch out of neurons and form the vital connections that allow signals to be transmitted within our brains. It is known that the levels of mitochondria in axons can decrease with age, and during the progression of neurodegenerative diseases.
Findings on Mitochondria Depletion and Protein Accumulation
Now, the team has found that the decrease of mitochondria in axons directly affects protein build-up. They used genetic modification to decrease the production of milton, a crucial protein in the transportation of mitochondria along axons.
They discovered that this resulted in abnormal levels of protein accumulating in fruit fly neurons, a consequence of the breakdown of autophagy, which is the recycling of proteins in cells.
By conducting proteomic analysis, they were able to identify a significant increase in eIF2β, a crucial part of the eIF2 protein complex responsible for the initiation of protein production (or translation). The eIF2α part was also found to be chemically modified. Both of these problems hinder the healthy function of eIF2.
Implications for Neurodegenerative Disease Treatment
Crucially, by artificially decreasing levels of eIF2β, the team found that they were able to restore the lost autophagy and regain some of the impaired neuron function due to axonal mitochondria loss. This not only shows that the decrease of mitochondria in axons can cause abnormal protein accumulation, but also that this occurs through an increase in eIF2β.
As people get older and neurodegenerative diseases become more common, the team’s findings are an important step in developing treatments for these serious illnesses.
Reference: “Axonal distribution of mitochondria maintains neuronal autophagy during aging via eIF2β” by Kanako Shinno, Yuri Miura, Koichi M. Iijima, Emiko Suzuki and Kanae Ando, 25 March 2024, eLife.
DOI: 10.7554/eLife.95576.1
This work was supported by a Sasakawa Scientific Research Grant (2021-4087), the Takeda Science Foundation, a Hoansha Foundation Grant, a research award from the Japan Foundation for Aging and Health and the Novartis Foundation (Japan) for the Promotion of Science, a Grant-in-Aid for Scientific Research on Challenging Research (Exploratory) [JSPS KAKENHI Grant Number 19K21593], NIG-JOINT (National Institute of Genetics, 71A2018, 25A2019), and the TMU Strategic Research Fund for Social Engagement.